Dimethylformamide - Energies ------------------------------------------------------------------------------------- Kimball Gamess/6-31g same,opt G94W/6-31g,opt Nucleus-Nucleus Vnn : 180.23058 180.23066 180.08970 180.08979 Electron-Electron Vee : 256.72510 264.77374 264.76824 264.76824 Nucleus-Electron Vne : -929.98466 -938.69044 -938.70460 -938.70460 Electronic energy Eel : -426.74952 -426.92614 potential energy V : -493.02898 -493.68604 -493.84666 -493.84667 kinetic energy T : 246.51004 246.87115 247.01022 247.01022 total energy Etot : -246.51894 -246.81489 -246.83644 -246.83644 Virial ratio -V/T : 2.000036 1.99977 1.99930 1.9993
Coordinates:
NC(CH3) Å: 1.4743 ¦ 1.4612 (avg.) NC(CHO) : 1.4743 ¦ 1.4137 CO : 1.2102 ¦ 1.2054 CH(CH3) : 1.1003 ¦ 1.0843 (avg.) CH(CHO) : 1.1003 ¦ 1.0900 OCN deg : 125.73 ¦ 124.30 HCH(CH3) : 112.39 ¦ 108.85 OCH(CHO) : 111.91 ¦ 119.60 CNC(CH3,CH3) : 106.36 (C's have ¦ 115.61 C(CH3)NC(CHO) : 106.36 equal param.)¦ 114.60
Dimethylformamide - Energies (Hartree)
-------------------------------------------------
One electron energy : -684.81120
Two electron energy : 257.85617
Nuclear repulsion Energy : 180.41847
-----------
Total Energy Etot : -246.53656
Nucleus-Electron pot.Energy Vne : -931.33467
Electron-Electron pot.Energy Vee : 257.85617
Nucleus-Nucleus pot.Energy Vnn : 180.41847
-----------
Total potential energy V : -493.06003
Total kinetic energy T : 246.52348
Virial ratio -V/T : 2.00005
Electronic energy Eel : -426.95503
Electron potential Vel : -673.47850
DMA2 - Interatomic Distances and Angles
NC(CH3) : 1.4716526 Å
NC(CHO) : 1.4699199 Å
CH(CH3) : 1.0987654 Å
CH(CHO) : 1.0727132 Å
CO : 1.2184562 Å
OCN : 122.57056 deg
CNC(CH3) : 106.71502 deg
C(CHO)NC(CH3) : 106.71502 deg
OCH : 113.50472 deg
NCH(CH3) : 106.70849 deg
HCH : 112.08696 deg
DMA2 - Cartesian Coordinates
12
N 0. 0. 0.
C 0.42326436 -1.409471 0.
C -1.4716526 0. 0.
H 1.5220297 -1.4093458 0.
H 1.0165492E-2 -1.8633589 -0.91138382
H 1.0165492E-2 -1.8633589 0.91138382
H -1.7875504 1.0523754 0.
H -1.7875504 -0.52618769 0.91138382
H -1.7875504 -0.52618769 -0.91138382
C 0.42276603 0.56837388 -1.2879769
O 1.1483951 1.543931 -1.3678916
H 0.12947952 0.17406669 -2.2415064
------------------------------------------------
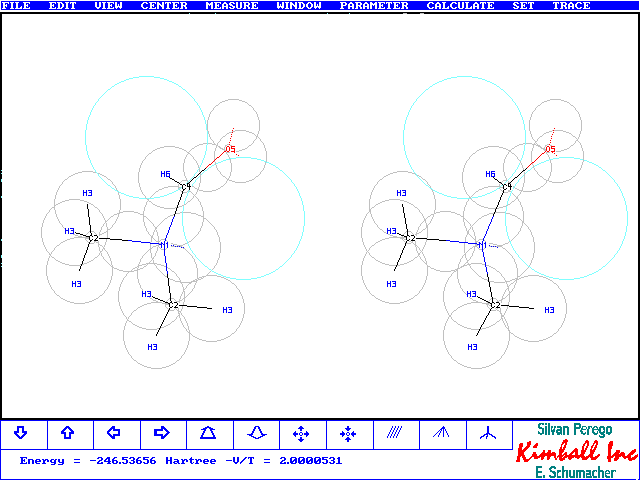
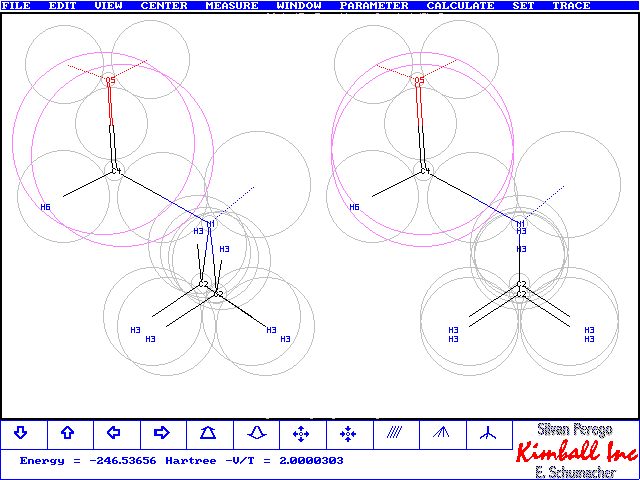
This Trace file from Kimball.exe comes from a second run of DMA which created the figure at the top. The numbers are slightly different from the comparison table above. In this run C4's distance from N1 has been variied independently from the N1C2 distance. Furthermore the
p-clouds were allowed to move along the CO-bond. This created 22 variables for the optimization and a slightly more stable result. Several distances and angles have changed a bit. However the N1C4 bond has not moved near the much lower value of the ab initio optimized structures. The reason is, that Kimball presently cannot model well
the partial NC4 double bond character of the NC=O moiety. It gives, however, correctly the planar arrangement of NCHO. The figure at bottom contains the numbered classes, referred to, and shows a better view on the NCHO region. The p-bond has moved nearer the C4 core and not, as perhaps expected, towards the more electronegative O core. It appears that the nearby N-atom and repulsion by the two compact oxygen lone pairs make up for this result. At the next step an sp2 N-Atom with two pz electrons would share in a three-center 4-electron p-bond system with C and O. This would generate a planar(C2)NCO moiety as obtained with most ab initio calculations, see the discussion on p. 105/106 in "Exploring Chemistry with Electronic Structure Methods", 2nd ed., J.B. Foresman & Ae. Frisch, Gaussian Inc. Pittsburgh, 1996.- Only the most exact model chemistries with RHF-SCF give non planar geometry, as known to be true from experiment. Interestingly Sto-3G monitors a transition state for the planar arrangement. Kimball is, of course, too much nonplanar as expected from a pyramidal N-node with the lone pair at one corner, see figure below.
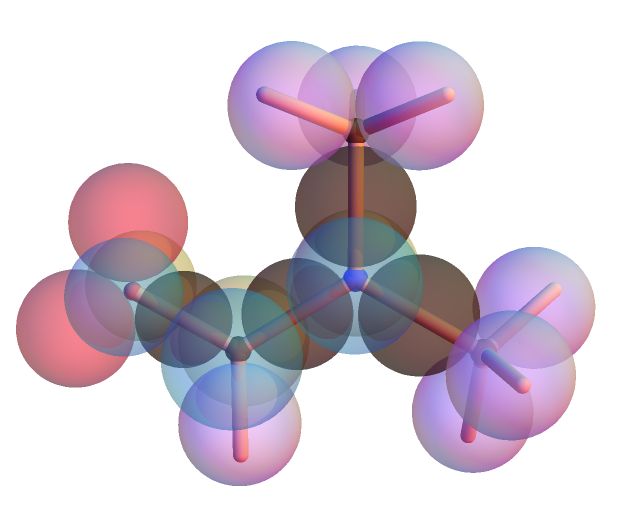
and here is a Kimball model of DMA from Mathematica-9 notebook: The five "heavy" atoms are all in a plane