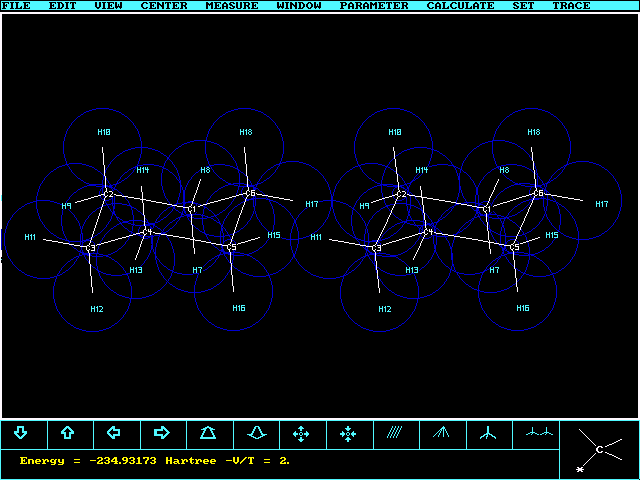
 cyclo-Hexane with chair conformation has been generated
and optimized with Kimball.exe (G2Fix.prf
prefactors,
containing some correlation energy) using 7 variables: Three Kimball radii
(C1s, C-C, C-H), three angles (CCC, HCH and dihedral HeqCCC), and the C-H distance.
Axial and equatorial C-H bonds are assumed to be equal (could be changed).
cyclo-Hexane with chair conformation has been generated
and optimized with Kimball.exe (G2Fix.prf
prefactors,
containing some correlation energy) using 7 variables: Three Kimball radii
(C1s, C-C, C-H), three angles (CCC, HCH and dihedral HeqCCC), and the C-H distance.
Axial and equatorial C-H bonds are assumed to be equal (could be changed).
The program finds, for the given tolerance, an energy minimum with C1 symmetry which
is within 0.001Å and 0.2 deg of D3d symmetry.
The total energy is slightly better than that of an SCF-calculation
in the Sto-3G -231.48267 or HF/4-21g basis -233.71719, and
similar to a HF/6-31g, -234.11102 or a
HF/6-31+g(2d,p) computation: -234.23538. The latter four are
based on optimized structures with Gaussian98 (symm=loose keyword) starting from
the given Kimball structure. Only the last finds the correct symmetry group D3d.
For Kimball.exe the time from the end of the graphical setup of the
molecule to reach the final result is 12 sec on a Pentium 233 MHz. The stereoplot
shows the numbering of the atoms during the construction with the node shown in the inset,
lower right corner: C1®C2®
C3®C4®C5
®C6, and ring closure C6®
C1. The H-atoms are added automatically.
Mopac7 finds a formation enthalpy at 298K of -35.7 kcal/mol with the AM1 hamiltonian
(exp. -29.4) and "Gradients were initially acceptably small, SCF field reached" using the
Kimball structure.