{kind=link}

Kimball.exe braucht, verglichen mit semiempirischen quantenchemischen Programmen, eine
sehr geringe Zahl Parameter. Diese sind anhand von ab initio Rechnungen bekannter Moleküle
geeicht worden. Dazu gehörten CH4, C2H6, NH3,
N2H4, CH3NH2, H2O, H2O2,
CH3OH, HF, F2, CH2F2, sowie C2H4,
O2 und HCHO für Verbindungen mit Doppelbindungen. Der Parametersatz, der in
den folgenden vier Bildern gezeigt wird, ist noch unvollständig und bei gewissen
Einträgen nicht genau (nur eine Stelle nach dem Komma). Es besteht also noch ein weiter
Spielraum, das Verhalten des Programms zu verbessern. Z.B. finden Sie für die Moleküle LiH, BeH2, bis HF eine mit Hilfe der Gaussian98-G2/G3 Energien
verbesserte Parametrisierung, welche zeigt, wie weit man etwa gehen kann. Ferner ist für gestaffelte Kohlenwasserstoffe mit der Eichung an den ersten paar
Gliedern eine übertragbare Parametrisierung erreicht worden, welche bis etwa C50H102
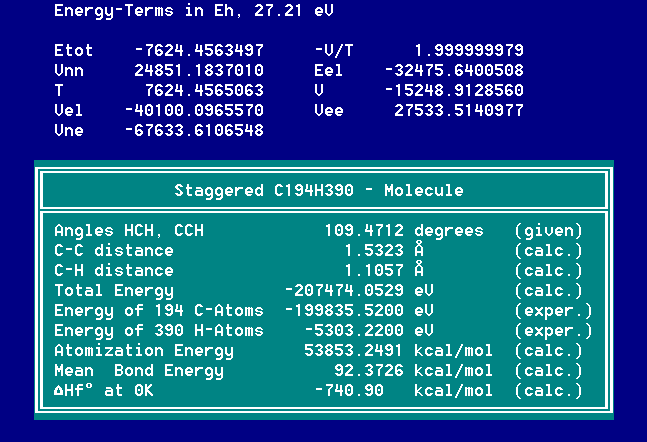
Bildungsenthalpien in wenigen Sekunden auf zehntel kcal/mol genau berechnen lässt.
Selbst bei C194H390 wird in 81 s (1GHz PIII) die folgende Bildungsenthalpie erhalten, welche auf 1 kcal/mol genau ist.
Die Parametrisierungskonstanten heissen "Vorfaktoren" (prefactors), weil der
unveränderte Faktor 1.0 jeweils dem nicht parametrisierten Kimball-Modell entspricht,
siehe Einführung. Der Faktor 1.0 ist eingestellt, wenn Sie das
Programm starten. Erst wenn Sie ein bereits gerechnetes Molekül oder eine
Vorfaktorendatei mit FILE / Load Molecule oder FILE /Load prefactors laden,
haben die Tabellenfelder evt. andere Einträge als den Faktor 1.0. Die vier folgenden
Tabellen entstehen, wenn Sie die Parameterdatei G21205.PRF laden. Versuchen Sie auch
G2Fix.PRF, eine noch unvollständige neuere Parameterdatei (mit immer noch konstanter
Abschirmkonstante) oder den früheren "Standard" ES0706.PRF.
Unter dem Befehl SET können Sie diese Tabellen selber abändern und damit eine Sie
interessierende Klasse von Molekülen präziser berechnen als es mit den Standard
Parametern möglich ist. Bitte speichern Sie solcherart veränderte Vorfaktoren unter
einem neuen Namen FILE / Save prefacors ab, wodurch sie Ihnen immer wieder zur
Verfügung stehen. Die Autoren
sind sehr daran interessiert, Ihre verbesserten Parameterdateien erhalten und in Zukunft
mit dem Programm ausliefern zu dürfen.
Kinetische Vorfaktoren (Einfachbindungen):
p - s kinetische Vorfaktoren (Doppelbindungen)
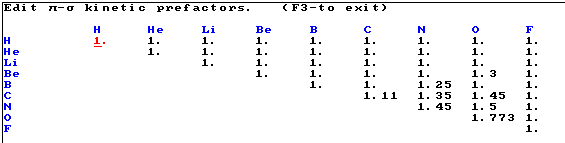
p - p kinetische Vorfaktoren (Doppelbindungen)

Kinetische Vorfaktoren für einsame Paare empty-c., zentrale 1s2
Wolken central-c., sowie
Vorfaktor e-e für die Elektronenabstossung in einer doppelt besetzten Wolke

Der letzte Wert der obigen Tabelle betrifft die Elektronenwechselwirkung innerhalb einer
Kimball Kugel. Gegenwärtig ist in Kimball.exe ein einziger Wert dafür vorgesehen. Es
zeigt sich, dass dies die grösste Fehlerquelle des ganzen Verfahrens ist. Man muss jeder
Art Kimballkugel, also z.B. der CH-, NH-, CC-Kugel etc. einen leicht verschiedenen Wert
zuordnen, um die Präzision zu erreichen, wie sie für die oben angegebenen, weiter
entwickelten Anwendungen, gilt, siehe unter Weiterentwicklung, 2. Dieser Ausbau des
Programms ist gegenwärtig in Arbeit.
Im Programm CHEMUNT \ Kimball \ en.exe, das die Elektronegativitäten der Elemente bis
Cr "ab initio" bestimmt, ist bereits gezeigt worden, wie die Erweiterung
vorgenommen werden kann. Die einfachste Möglichkeit ist ein zweistufiger Vorgang. Man
rechnet vorerst jedes Atom der zweiten Periode mit dem He- und Ne-Rumpf. Hernach werden
die 8 Elektronen des Ne-Rumpfs gemeinsam als Kugelschale beschrieben mit gleichgrosser
kinetischer Energie wie die 4 doppeltgefüllten Wolken über dem He-Rumpf. Wenn dieser
Rumpf nun in einen Verband eingebracht wird, zB H2S oder PH3, so
wird er nur mit einem (zu optimierenden) Radiusparameter für die Ne-Schale
behandelt. Dies hat Ähnlichkeiten mit der "frozen core" Näherung
verschiedener quantenchemischer Methoden, z.B. GAMESS oder Gaussian94. Auf dieses kugelige
Gebilde werden nun die Elektronenwolken der zweiten Periode genau gleich aufgebaut wie
jene der ersten auf den He-Rumpf. Man wird dadurch von einer viermal grösseren Zahl von
Variablen verschont und ausserdem von der etwas unphysikalischen Problematik, wie man nun
die Aussenkugeln zweckmässig in die "Vertiefungen" der inneren, vollständigen,
Schale verteilen soll. Dieses Verfahren kann nach Erreichen der Argonschale wiederholt
werden und gibt bis Chrom sehr gute Werte. Darüber hinaus wurde es noch nicht geprüft.
Diese Erweiterungen müssen noch ins Kimball.exe Programm eingebaut werden.
Für die Elektronenabstossungsenergie Vee in einer gemeinsamen Wolke wird der Wert 6/5R*(N -(N/2)1/3) hartree, R in
bohr, N Anzahl der Elektronen, berechnet. Mit N=2 erhält man also gerade 6/5R, wie von
Kimball als elektrostatische Wechselwirkung zweier superponierter Ladungskugeln gleicher
Dichte mit je einem Elektron bestimmt. Jedes Elektron wird vom anderen von der Ladung des
im Zentrum der Kugel sitzenden Kerns "abgeschirmt", wodurch nur noch eine
effektive Kernladung wirkt, welche um die "Abschirmkonstante" vermindert ist.
6/5R ergibt eine Abschirmkonstante von 0.4 Ladungseinheiten. In der Einführung
steht mehr zu dieser Problematik. Hier sei nur das Desideratum festgehalten, dass auch
diese Grösse einen (empirischen) Vorfaktor erhalten muss. In der jetzigen Programmversion
ist nur eine mittlere Abschirmungskonstante, die gleiche für alle doppeltbesetzten
Wolken, enthalten. Beim Laden der Vorfaktorentabellen wird s =
0.3354 (entspricht ca. dem Rumpf des N-Atoms) gesetzt. Bei J.C. Slater (1930) war diese
Konstante 0.3 für Li ... F während E.Clementi & D.L. Raimondi (1963) eine neue
Tabelle von Abschirmkonstanten vorschlugen, in welcher die Werte einzelner Atome
verschieden gross sind. Von Li, Be, ..., F ändern sie sich von 0.3094, 0.3125, .., bis
0.3499. Versuche haben gezeigt, dass die Präzision der
Kimball-Rechnungen erheblich verbessert wird, wenn s einer
Parametrisierung unterworfen wird.
Besonders notwendig ist diese Anpassung im Falle der Kugeln, welche ein Proton enthalten,
also bei gebundenen Wasserstoffatomen. Es gibt eine "tiefere" Begründung dazu,
welche die Dichtespitze beim Proton bei der Bestimmung der Korrelationsenergie einbezieht.
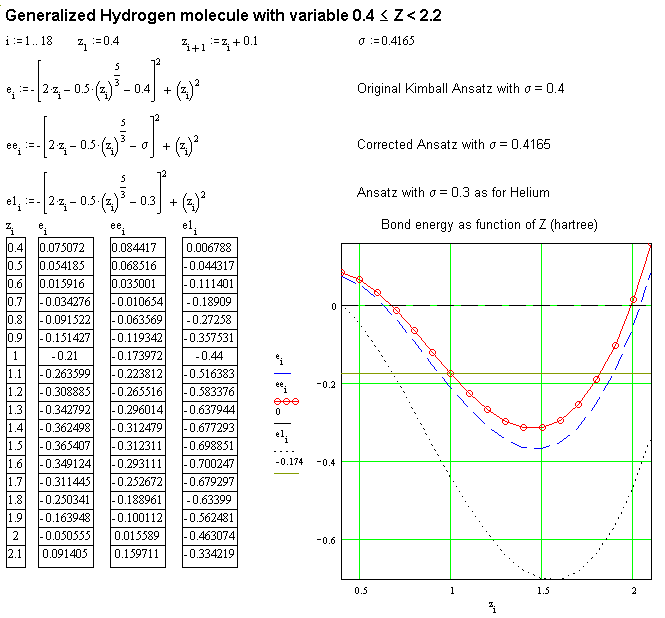
Beim Wasserstoffmolekül gibt es zwei Dichtespitzen in der gleichen Kimballkugel, wodurch
der Vee-Wert statt 6/5R 6.2475/5R oder s = 0.4165
erreicht, wie die folgende Figur zeigt:
Verbesserung des Frontends, der Stabilität (weniger Abstürze bei schlechten
Startwerten...). Vereinfachung der Struktureingabe, Windowsoberfläche, C/C++ oder Java
statt Modula-2 Sprache. Bessere Behandlung der Delokalisation. Fähigkeit, freie Radikale,
angeregte Zustände (ausser He(1s2s und He(1s2p)!), zu berechnen. Ausgabe von
Bildungsenthalpien bei 0 K. Verbesserung der Bestimmung der Schwingungsspektren (und
thermodynamischen Funktionen), NMR-Abschirmkonstanten. Erweiterung auf Kristallgitter
(siehe Diamant und Li-Metall).
Die meisten dieser Desiderata sind bereits in unabhängigen Programmen realisiert. Sie
müssen nun noch in das Kimball.exe Programm integriert werden.
Letzte Änderung 27. Feb. 2002 ES