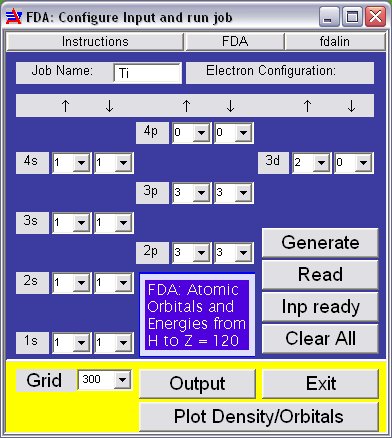
 This little script lets you compute energies and orbitals of atoms for any electron configuration you fancy: Atoms in groundstate, atomic ions, ionization potentials, electron affinities, inner excitations for X-ray terms, outer excitations for UV/VIS spectra. The various buttons on the FDA panel are explained here. You can fill the boxes for composing a special electron configuration and run it with [Read], or use [Generate] with an atom symbol to refer to the stored ground state configurations (which can be edited before computing). [Inp(ut) ready] lets you run a previously generated input file.
This little script lets you compute energies and orbitals of atoms for any electron configuration you fancy: Atoms in groundstate, atomic ions, ionization potentials, electron affinities, inner excitations for X-ray terms, outer excitations for UV/VIS spectra. The various buttons on the FDA panel are explained here. You can fill the boxes for composing a special electron configuration and run it with [Read], or use [Generate] with an atom symbol to refer to the stored ground state configurations (which can be edited before computing). [Inp(ut) ready] lets you run a previously generated input file.The density and orbital arrays can be plotted with [Plot Density/Orbitals] (see below).
The computing engine FDA is based on an SCF procedure proposed by Herman & Skillman (1963) using Density Functional theory and the Xalpha functional. The source code from David J. Heisterberg (1995) is an improved and corrected implementation and FDA gives significantly more accurate results. It may be downloaded as fda.tar.Z. It is written in C/C++ and can be compiled e.g. with Visual C/C++, 6.0.
We have compiled the program, bundled it with the script fdalin and test examples as
fdalin.zip. Download for
Windows9x,ME,2k,XP,Vista,7 and for Linux (using the wine emulator Version 1.x.x). Clicking the buttons [FDA] and [fdalin] in the top menu bar of the panel gives you a short introduction by David J. Heisterberg's FDA and to some examples of using this script. FDA is freeware but must carry Heisterberg's copyright.
Notice: The model is not spin polarized and non relativistic. Overall, it has an accuracy between LDA (local density approximation) and LSD (local spin density approximation). The following pictures from NIST show the accuracy of these and other DFT methods across the PS for ionization potentials and the expected corrections of the atomic energies resulting from spin polarization.
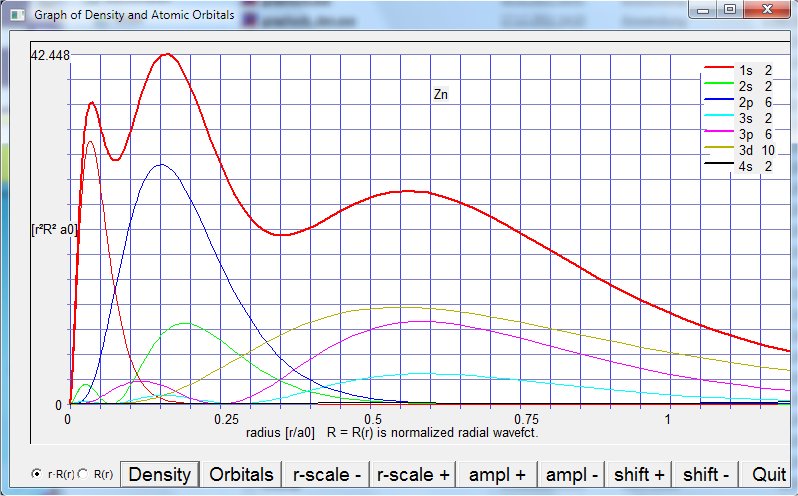
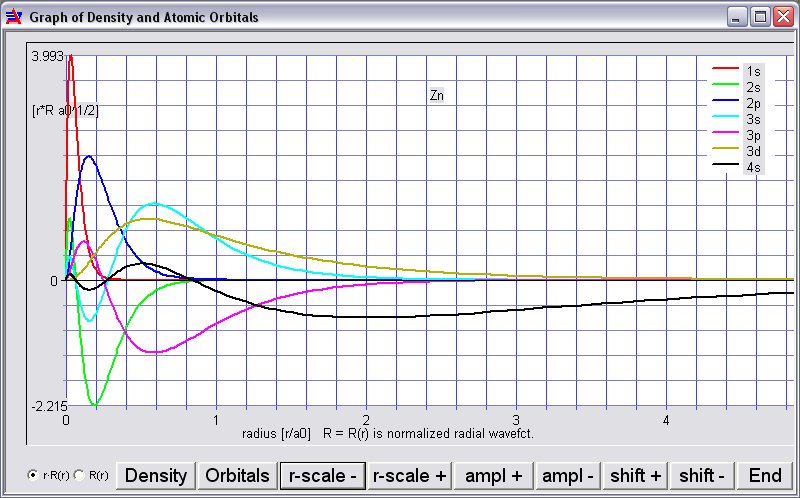
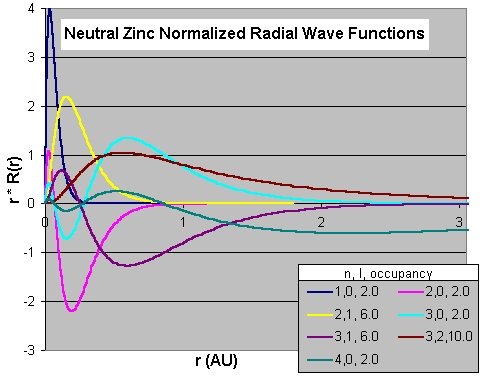
Output window for neutral Zinc (summary) using [Plot Density/Orbitals]: