|
First Principles Methods: The name implies that there is no calibration done with empirical data. The huge archives of basis sets which are constituents of trial wavefunctions have certainly been optimized with sets of experimental data. However, this is not parametrization, because it just helps to give the trial wavefunctions a flexibility to better and better approximate the unknown true wavefunctions of a molecule, all guided by the Variation Principle. It turns out that the linear combination of Atomic Orbitals to construct Molecular Orbitals is often not the best choice. It can be defended because the complete set of spherical AOs represents an orthonormal set which asymptotically can approximate every (well-)behaved function, hence also an MO. The usual chemists' jargon with LCAO-MO arguments, however, truncates these infinite expansions at the first term(s) which is(are) far from any "optimized wavefunction". At the beginning of QC, computers were not capable of doing much better. Today this has changed dramatically. Fast computers have also opened a way to use other orthonormal function sets: One of them are planewaves, e.g. eikr, cosine and sine functions. The famous Car-Parrinello code as realized in the CPMD program package, entirely works with these. The method makes use of the existence and applicability of algorithms for Fast Fourier Transformation FFT. Together with Pseudopotentials, summing much of the contribution of low lying core orbitals and with the help and advantage of DFT functionals this has become the method of choice to follow dynamic trajectories even of large chemically reacting systems. It is amazing how much we have learned from this method in the last 20 years. Chemists may wonder when they see familiar energy level and orbital surface plots which are not derived from atomic orbitals, but computed from planewaves: The picture shows a density surface for B2H6, computed | with more than 500'000 plane waves in 3 min on my home computer. Since 1960 (S.Golden, Rev.Mod.Phys. 32(322-327), and 1987 (W. Yang, Phys.Rev.Lett.59,1569-1572) DFT functionals without any orbitals have been developed. So, why stick to atomic orbitals for representing molecules? 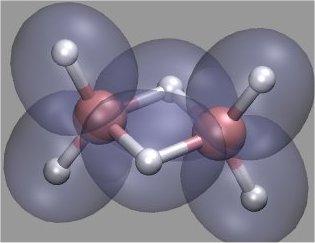 Every molecule is a new entity and not (accurately) derivable from its atoms - atoms are not bricks, molecules not houses (none of the many schemes to parse molecular electronic density into atomic contributions can be realized without arbitrary assumptions. Deep down, NMR shows more and more atomic character because a large part of the interaction with nuclear spins happens in a fairly shielded assembly of spherical <ns2> core densities).- In the last 20 years John Pople and his crew (citaton), and later without him, have developed the combination QC methods Gaussian G1, G2, ... ,G4. The aim is to reach "chemical accuracy" for enthalpy of formation, ionization potentials, electron and proton affinities. This has been an heroic undertaking. I've followed and used it carefully, because I was applying this for parametrizing Kimballs model. The methodology made great progress and is now within about 0.5 - 1.0 kcal/mol of experimental formation enthalpies with excellent predictive capability. But, a final correction determined with a large basis of experimental data, i.e. parametrization called "highher level corrections" in Gaussian09™, is still necessary. For CH4 it amounts to about 1/4 of one CH bond energy! G3MP2 is now offered in Gamess as well. Every molecule is a new entity and not (accurately) derivable from its atoms - atoms are not bricks, molecules not houses (none of the many schemes to parse molecular electronic density into atomic contributions can be realized without arbitrary assumptions. Deep down, NMR shows more and more atomic character because a large part of the interaction with nuclear spins happens in a fairly shielded assembly of spherical <ns2> core densities).- In the last 20 years John Pople and his crew (citaton), and later without him, have developed the combination QC methods Gaussian G1, G2, ... ,G4. The aim is to reach "chemical accuracy" for enthalpy of formation, ionization potentials, electron and proton affinities. This has been an heroic undertaking. I've followed and used it carefully, because I was applying this for parametrizing Kimballs model. The methodology made great progress and is now within about 0.5 - 1.0 kcal/mol of experimental formation enthalpies with excellent predictive capability. But, a final correction determined with a large basis of experimental data, i.e. parametrization called "highher level corrections" in Gaussian09™, is still necessary. For CH4 it amounts to about 1/4 of one CH bond energy! G3MP2 is now offered in Gamess as well.
| |