{kind=link}
N.Moll, G.Meyer,
Nature Nanotechnology 7,
227-231(2012)
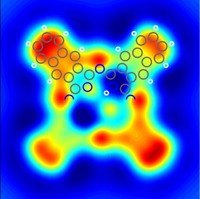
on surface by a
combination of
scanning force microscopy
showing charge distribution
of "orbitals"
EnvironmentThis K-Model starts with the observable three-dimensional electron density. It is a simple forerunner (akin to Slater's X-alpha method) of Density Functional Theory (DFT). It uses real space spherical elements | "orbitals" and curly arrows to move them around in order to visualize and "mechanistically" interpret and predict chemical reactions. This is a great success and nobody cares whether the charge domains are called "orbitals", "electron domains", "electron clouds" or "(electron)pairs" (as two dots or one dash). There are many empirical facts and several rules to learn in order to master this art and to profitably talk organic chemistry with their help. All this is not directly derivable from the quantum chemists "orbital" concept. It is an inventive profane name pick up, perhaps identifiable with the "chemical orbital" <½*½> friendly conceded by R.S.Mulliken, which we often imagine to be "seeing" on the screen of a scanning tunneling microscope when a tip is hovering over a molecule: |
|
F.Mohn, L.Gross, N.Moll, G.Meyer, Nature Nanotechnology 7, 227-231(2012) |
| Naphthalocyanine on surface by a combination of scanning force microscopy showing charge distribution of "orbitals" |
FocusVery likely, it was Kimball's intention (I presume) to enhance this qualitative success-story, which began shortly before WW II, by a quantitative aspect. It seemed just manageable by the computers of 1950 to replace the various shapes of artwork "orbitals" by spherical electron "clouds", the simplest 3D-domains. The aim was to obtain useful ground-state energies and equilibrium structures of molecules by a minimal calculational effort. An essential first step then was to approximate the kinetic zero point energy of such a sphere in order to obtain the correct quantum-mechanical "electron pressure" to counterbalance electrostatic interactions. Although there have been several attempts to derive the K-model from quantum mechanics, none of these is correct (e.g. [1], [2] both falsified [3]). Some properties of the hydrogen atom may be an exception: Without explicitly solving the Schr—dinger equation, the kinetic zero point energy of the H(1s) groundstate is derivable from an argument with Heisenberg's uncertainty relation, for which G.E.Kimball (1951, G.F. Neumark, loc.cit.) found | Now proceed with this as follows:
If you want to extend this terse description, see the Tutorial or read [7]. |
|
[1] F. Rioux, J.Chem.Educ. 50(1973)550 [2] F. Rioux & P. Kroger, Am.J.Phys. 44(1976)56 [3] Ch.K. J½rgensen, Chimia 31(1977)445 [4] H.Hellmann, Quantenchemie, p.9(2,7), p.42(9,5), F.Deuticke, Leipzig & Wien, 1937, and [13], p.49(3.1.9), p.108(6.1.19) | [5] S.F.Boys, Proc.Roy.Soc., 200A(1950)542 [6] JCP 47(1967),3707,3714 [7] J.P. Platt, "Chemical Bond and Distribution of Electrons in Molecules", Hdb.der Physik, Vol. 37/2(1961) p. 258 |
First qualitative caricatureHenry Albert Bent (HAB for short) has been inspired by Kimball and from 1963 published several didactically remarkable articles (mostly in J.Chem.Ed., e.g. [8], [9]; see a history of this in [10]), wherein he has drawn many chemically interesting conclusions from a "tangent sphere model", a qualitative interpretation and restriction of the model which Kimball explicitely did not subscribe to. One important conclusion from HAB's work is, that the VSEPR model of Gillespie-Nyholm-Sidgwick (also qualitative) and now a worldwide epic in chemical education for the prediction of molecular structures, is often chemically less accurate or even misleading in comparison to Bent's approach. Bent has just compiled his life-long endeavours in a new book [11], where he stresses his points of view, now | condensed into a nonmathematical, nonphysical but very intuitive (i.e. noncommunicable) "Conceptual Valence Bond Theory". - (Comment: In my opinion, much of this is just rewording well known facts and concepts into a new guise without new insights.- It is amazing how much HAB misunderstands and ignores the results of a field which has lived to see such a triumphant progress during the last 30 years: first principles Quantum Chemistry, incl. DFT! Bent argues with the disputes of the quantum chemists between the thirties and fifties of last century, when H2 and He were among the few systems amenable to high quality calculations and the rest of chemistry a bag of colorful "theories" about which their protagonists fought fierce battles, settled long ago. A readable summary of this history is in [12]: 'Hyperbola of Quantum Chemistry', or in the Nobel Lecture of Walter Kohn, 1998). |
|
[9] H.A.Bent, J.Chem.Ed., 45(1968)768 [10] E. L. Schultz, J.Chem.Ed. 63(1986)961-965 |
[12] B. S. Park, Annals of Science, 60(2003)219-247 |
RestorationIn what follows here, many examples show, that there is no reason that Kimball's quantitative spherical clouds of a molecule should be tangent over and beyond those between adjacent atoms (comment). They may often be (nearly) so if one actually does the calculation, e.g. in SF6 or in C2H6. SiH3CH3 is a mixed case. With other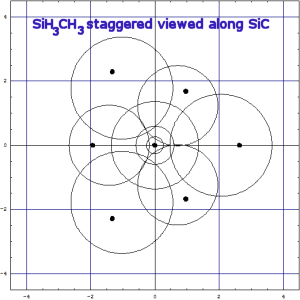 | 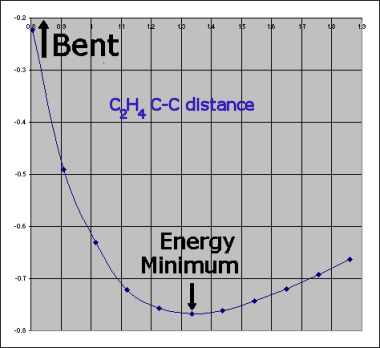 (And, yes HAB: of course, we know that proper π-bonds have an electronic angular momentum around the bond and should only be named "π" for molecules where that bond direction is coincident with a C∞ rotation axis, e.g. diatomic molecules, HCN, HCCH, or O=C=O, but not for H2C=CH2 (inspite of all organic chemists and most theoreticians, who do so!)). |
Developments 1For a long time and with many on and offs (since 1960!), the present author has tried to explore the possibilities and limits for application of a quantitative K-model to molecular structures and other chemical problems. It turned out, that for all systems more complicated than the H-atom, parametrization is necessary because the K-model does not offer an application of the Variation Principle, although a molecular structure is found by a minimization of the energy. The Variation Principle is only available for the proper wavefunction treatments and is often not true for DFT methods | with approximate functionals. Many good DFT functionals and basis functions, e.g. B3LYP/6-31G, give an H2 molecule
which is more stable than nature has it! see [13]. The good news is, that Kimball's Ansatz can be parametrized by experimental data and ab initio quantum-mechanical results, and, the parameters are transferable. This means that parametrizing e.g. CH4, NH3, H2O, HF, C2H4, C2H6 (and other small molecules) allows the computation of whole classes of compounds like hydrocarbons, amines, aldehydes, ketons, carbonic acids, aminoacids, polypeptides, carbohydrates, aromatics, heterocycles (for DNA) and many inorganic compounds as well, see gallery. |
| [13] R.G. Parr & W. Yang, "Density-Functional Theory of Atoms and Molecules", Oxford Univ. Press, Oxford, 1989, (ISBN 0-19-504279-4), p. 52 ff. |
Developments 2Dr. Silvan Perego has developed a program Kimball.exe during his diploma thesis with me (University of Bern, 1989). It is an approach to a parametrized quantitative Kimball-Model. A small set of parameters is sufficient to obtain the precision of force-field methods for molecular energy and structure. In comparison to first principles ES-methods it produces energies and structures near RHF/6-31g model chemistries as computed with Gaussian98,03 or Gamess. Presently, Kimball.exe is limited to molecules and ions with up to 50 atoms from | Here is a short animation of using the program to build and compute H2. It shows, that setting up a Kimball calculation is straightforward: Just draw a Lewis sructure of the molecule and replace every valence stroke (and the 1s cores!), including lone pairs, with a spherical cloud. Their radii are all different, except for symmetry equivalent atoms. Now sum the kinetic energies and include all classical electrostatic interaction energies +/-, +/+, and -/-. Identify free angles and dihedrals as additional variables. Then a start value is allocated to every radius and structural variable and the minimum energy sought by repeatedly improving their values with several minimization algorithms until the total energy converges to a given small fraction near its (local) minimum. This is, essentially, what Perego's program does, see table. If you look into one or the other Wolfram Mathematica™ examples, below, you will see this explicitely. A good FindMinimum[function] of many variables is part of Mathematica's arsenal of procedures. |
Developments 3Beyond the limits of Perego's Kimball.exe, Wolfram Mathematica™ programs treat atoms, ions, molecules and crystals from H to Xe and Cs, and molecules of several hundred atoms, see the list, sort of work in progress. Higher multiplet states have also been tested successfully as well as excited states. The problem of non-local electron states is solved in certain cases by filling orbitals with only one electron. E.g. Benzene's C(pz) type orbitals | allow overlapped "ringclouds" above and below the molecular plane, which produce the NMR detected diamagnetic shielding by a "ring current". It is also easily possible to model 3D-electron clouds mimicking |
Results and more to read and contemplateKimball's atoms are equivalent to those computed with Slater-type-orbitals. |
|
Technical details of K model computations |
Critique of Kimball's ModelGeneral: 1) A very harsh verdict against Kimball's model can be read in the book of Hans Primas & Ulrich M■ller-Herold, 1984, [14], where they qualify the model simply as trash (Schund), without saying what it is nor giving a reason.2) Ch.K.J½rgensen [3] criticizes the papers by F. Rioux [1],[2]. He mistakes Rioux' incorrect derivation of the kinetic energy of the electron in the H-atom for an introduction to Kimball's model (mistake corrected in next issue of Chimia, when informed by a referee).- If there are more criticisms in the literature, none has been found so far, a clear indication, that Kimballs model has never been accepted nor dismissed as result of a serious scientific analysis. |
Specific: Critique of the model and its methods:
|
| [14] Hans Primas & Ulrich M■ller-Herold, "Elemente der Quantenchemie", Teubner, Stuttgart (1984), page 315 |
Summing upIt remains to you, the reader, to decide whether a quantitative K-model (distinct from the qualitative caricatures) serves any purpose in your endeavor to categorize chemical insight! Models are never true, but they may be useful ... and fun to develop, see e.g. the recent artwork (is it science? another Kimball caricature?) Kimball-Model in chemistry didactics at ETH, Z■rich. Compare the last picture of a K-atom with the quantitative K-atom computed in 1963 by Willy Heinzelmann (Univ. of Zurich). Correct representations are now the spherically symmetric atoms e.g. K and Cs, still truly "Kimball" computed.I do not claim, that there is anything new on this page. Most ideas are already expressed in the five theses under the supervision of G.E.Kimball. It has become a bit easier today to realize them, because computer resources are tremendously better than in 1950-1957. This has allowed to remedy weaknesses of the model by parametrization. In addition, the scope of the model, which many adepts of its qualitative interpretation have arbitrarily fixed at Neon or | Potassium, has been pushed to the second row of transition metals and up to Cs. There is no limit to go to the Lanthanides, Actinides and beyond. Rather, time has become short to do so! However, it is always beneficial to investigate how much mileage you get from a simple model (in US slang). The ill fate of Kimball's model: It was too early. 1952 no chemist had a desktop computer. Now it is too late, because he has black-boxes, ready to give answers with very little thought input of the user. This leaves our kids in school unprotected from questionable methodology: Misuses of quantitative models as qualitative and ununderstandable "Quantum Chemistry brought to the Highschools", both with no educational value. Kimball's quantitative model has the merit of being falsifiable and sometimes really fails on account of its simplicity. What a healthy experience for a young mind! In contrast: Most textbooks of chemistry ignore e.g. the abundant failures of the electron pair bonding model of G.N.Lewis and thus disqualify it as a scientific object! Never teach it as gospel! Stimulate scientific discussion about its merits and where in chemistry it fails. |
EpilogueWhy should anyone be interested in such a simple and, admittedly, crude model of the electronic structure of matter - 60 years of age -, if you need a computer to handle it, anyway ? Out there are tons of freely (gratis!) available modern software packages of a much better quality: Just google for Gamess, Firefly, NWChem, Orca, EHT, Mopac, CPMD, (and commercial ones like Gaussian09, TurboMole, Schr—dinger, Dirac, | Molpro, QChem a.o.) which also run perfectly well on modern homecomputers ? And, there are many good programs for extracting chemical information from these "numbercrunching" engines, some with exceedingly well done graphical interfaces, like Molekel, Molden, WebMO, VMD, gOpenMol, PCModel, RasWin, Ghemical, ArgusLab, Mask, Avogadro, MacMolPlt and many more. |
Perhaps,you are a freshman student and try to understand, what you are doing ?or a teacher, who wants to start his pupils with a simple microscopic (atomistic) view on chemistry, to be refined when more experimental facts are known which lead to deeper questions and demand better explanations ? or an organic chemist who would like to have a simple frame for daily work, quickly checked on a desk computer and helping to pep up Lewis dashes and dots - nearly 100 years old ? Most scientists use simple models to develop an idea! |
BonusRest assured, that using Kimball's model does no harm to your mental health nor hamper your entry into the "good science" of quantum chemistry. On the contrary! You will find many basic ideas, learned with the model, in refined shape: All the energy sums over a finite molecular space of Kimball's model are again there, but become integrals over complex functions in an infinite (actually: truncated Hilbert-)space - yes, that's the reason you need powerful computers. I can help you getting started e.g. with Gamess, even on your homecomputer! Ethene is computed in a breath, without bananas, but seemingly correct! |
| Acknowlegment: From 1960 to 1964 and, sporadically, in the next couple of years, I have had an active group of students who contributed some of their spare time to our understanding of the Kimball model. They used deskcalculators to find the real zeros of third degree polynomials to which all two variable Kimball problems can be reduced. Later, an IBM1620 (W.Heinzelmann, Univ. Z■rich) and various small HP computers were used to find energyminima of problems with more than two variables. At the Univ. of Bern in the late 1980, S.Perego and M.Hehlen have made important contributions. Several outstanding colleagues have been kind in tolerating (one of) my queer hobbies! | I thank all my students and am sorry that I have only been able to present here a few percent of their labor (all recently checked and recalculated, remaining errors my fault!). I did not publish any of this in the scientific literature for lack of time and courage.- I do not mention more names because one gymnasium chemistry teacher (not associated with me) has recently been fired by his university for presenting Kimball's model to didactics students. Myself, being beyond 20 years after retirement, am no longer afraid of getting fired ... Anybody may use this material, preferably cited (this URL), as long as (s)he is not laughing at me. |
| Last updated June 17, 2013 Page by ES. |
{kind=link}
{kind=link}
{kind=link}